Definition
The Western blot test detects proteins of the immune system and has been used since the late 1970s. Western blotting or immunoblotting can pinpoint one or more specific protein antibodies from a sample via a standard protocol. The Western blot protocol begins with the separation of larger molecules via electrophoresis. These denatured molecules are then blotted onto a specially-developed membrane. Tagged specific antibodies can then identify a particular antigen.
What is Western Blotting?
Western blotting is a reliable method of separating antigens from a mixed protein sample. Antigens are foreign substances that cause an immune response. Western blot results have high sensitivity and high specificity; this means there are very limited false-negative results and false-positive results respectively. In simpler terms, high sensitivity gives accurate results from samples with evidence of disease and high specificity identifies high rates in those who do not have this disease.
Even so, a Western blot result is not fail-safe. Antibodies may detect more than one type of antigen. For example, if someone is being tested for an HIV infection and, at the same time, is suffering from a lung infection, antibodies may pick up on the respiratory infection and fail to report the presence of HIV. Precision during each Western blot step is essential to increase test accuracy.
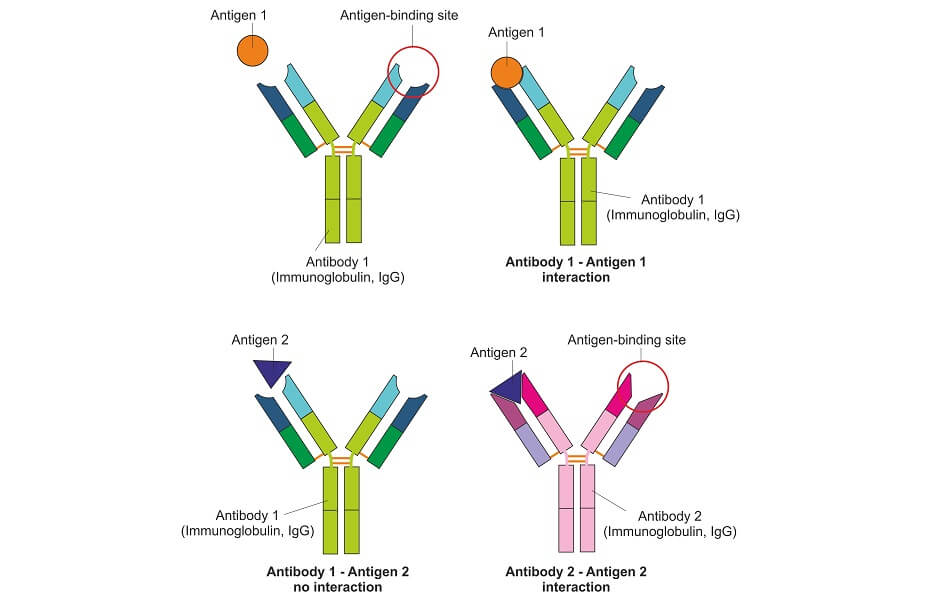
This test detects the presence of specific proteins within a mixed tissue, blood, urine, or saliva sample. It is used to detect disease, the effects of pharmaceuticals, and sports doping. It also has an important role to play throughout the fields of scientific and clinical research.
Western blot analysis is implemented in the field of molecular biology and named in reference to its predecessor, the Southern blot test, which detects DNA sequences in DNA fragments. Another similarly-named test is the Northern blot that pinpoints RNA molecules.

Western blot tests are carried out using the same steps; however, different products and techniques are used. These will be discussed further on.
Western Blot Steps
There are eight Western blot steps, all of which must be accurately performed:
- Western blot buffer preparation
- Sample preparation
- Separation of protein mixtures via gel electrophoresis
- Transfer of separated proteins onto a membrane (blotting)
- Membrane blocking
- Antibody incubation
- Detection of the target protein
- Data analysis
Buffer Preparation
Western blot testing requires various buffers that resist changes in sample pH during the testing process. Stains – Coomassie brilliant blue and Pinceau S, for example – and destaining solutions must also be mixed and diluted according to standard recipes.

Buffers are used during cell lysis (breakdown), protein separation, protein transfer, and blocking phases.
Sample Preparation
To carry out Western blot analysis, you first need a tissue sample. This sample also needs to undergo preparation. Sample cells are ruptured using mechanical methods at low temperatures. Low temperatures prevent proteins from degrading. Methods of cell disruption are:
- Homogenization: mixing particles to form an emulsion or solution
- Sonication: using sound energy (ultrasonic frequencies) to agitate and mix particles
- High-pressure disruption: homogenization under high pressure
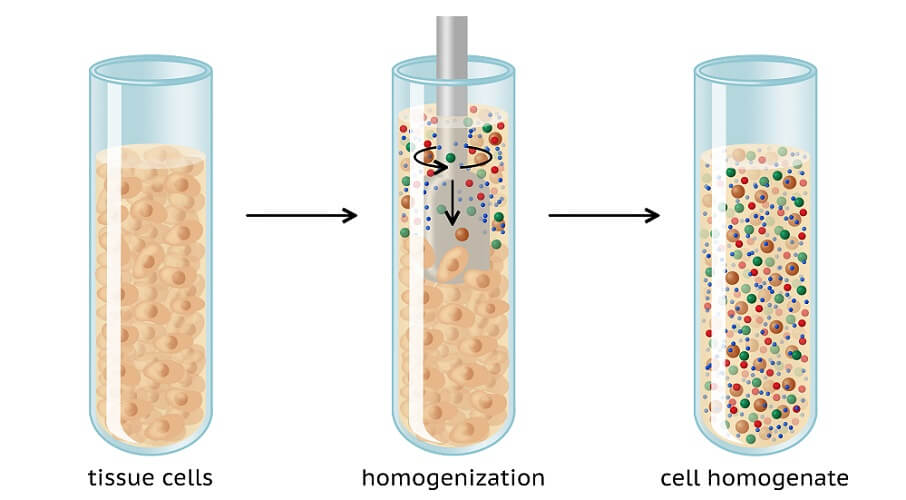
The sample is then further broken down with a buffer. The correct lysis buffer must be used or the to-be-studied protein may not be sufficiently extracted.
Protein Separation
Tissue-based samples are complex. They contain vast numbers, types, and structures of smaller polypeptide chains and larger proteins. They can also contain lipids and carbohydrates.
Every cell contains organelles constructed partially or primarily of protein, from microtubules to mitochondria. Detecting one protein type from any tissue sample is impossible without laboratory techniques such as Western blotting.

The next Western blot step is, therefore, to separate these macromolecules within a sample. While preparation ruptures whole cells, protein separation takes this a step further. The standard method of protein separation is gel electrophoresis. Gel electrophoresis uses an electrical current to force macromolecules through a gel slab.
A typical electrophoresis chamber is comprised of a gel slab sitting between two glass or paper plates. Spacers within the gel slab keep the chamber uniform. The thicker the slab, the more samples it can accommodate; however, heat will not dissipate as efficiently and this makes results less accurate. Electrophoresis chambers are placed in an electrophoresis tank that provides the necessary electric field.
There are two types of electrophoresis – one and two-dimensional. The one-dimensional method separates routine proteins. Two-dimensional electrophoresis allows you to compare multiple protein samples at the same time and on the same gel. Different target proteins are tagged with different color dyes. This article describes the one-dimensional form.
Native polyacrylamide gel electrophoresis (PAGE) and sodium dodecyl sulfate-PAGE (SDS-PAGE) are the most commonly used methods of protein separation in Western blot analysis. These use a single resolving gel. Another name for resolving gel is separating gel.
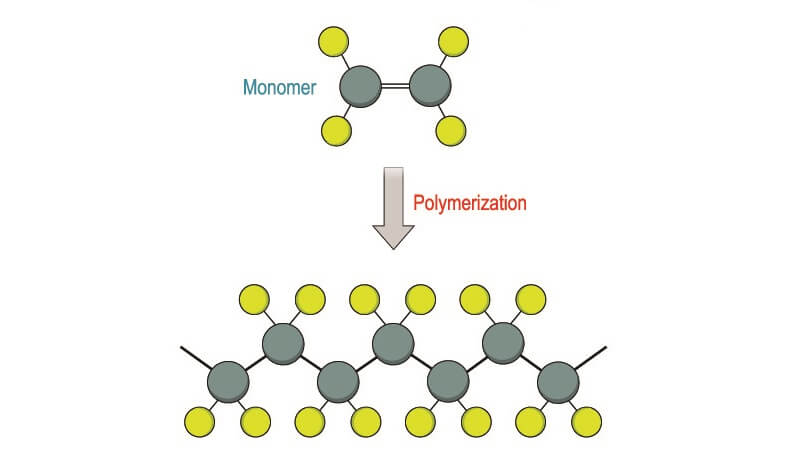
Other chemicals are added just before the test is carried out to polymerize (create molecular chains) within the gel. These are added without the presence of oxygen as O2 interferes with the polymerization process. This means that the electrophoresis chamber must first be degassed. Polymerization affects the size of pores running through the gel and makes the final Western blot result more accurate.
Another gel – a stacking gel – causes sample proteins to line up. Stacking gels act as the equivalent of the starting line at a horse race, lining up participants before they move (much more slowly) through the resolving gel.
As different proteins have different molecular weights, they move through the resolving gel and onto a membrane at different rates. This separates them from one another.
Protein or Western Blot Transfer
Protein transfer is the result of an electrical charge that surrounds the sample proteins. Within a controlled electrical field, proteins are led and pushed from gel to membrane. When the separated proteins reach the membrane, they ‘blot’ onto it.
Western blot transfer occurs either wet or semi-dry. Other, slower methods are diffusion, capillary action, and vacuum transfer. While wet electroblotting in an electrophoresis tank gives more accurate results, semi-dry techniques are quicker.
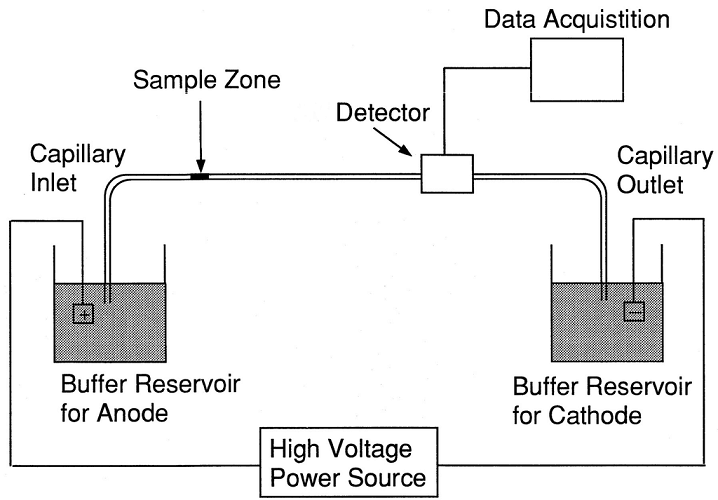
The blotting membrane sits closest to the positive electrode of the gel electrophoresis tank and charged proteins are pulled toward it. They are surrounded by negatively charged ions at the start of the protein separation phase. When an electric current is supplied, buffer chemicals push negatively-charged ions in front of and behind the proteins. These negatively-charged borders then move along the chamber at different rates, attracted by the positive charge close to the membrane. Speed is controlled by the molecular weight of individual proteins (molecular sieving).
The separated proteins finally blot onto the membrane. These protein stacks, sandwiched between negatively charged ions, create stained lines of various thicknesses according to sample quantity; they appear at different levels according to molecular weight. Pore size affects how many proteins move through the gel slab – larger proteins are blocked by smaller pores.
It is possible to purchase electrophoresis chambers with pre-marked proteins of which the accurate molecular weights are known. These are called calibrating gels and used as reference markers to determine the molecular weights of lesser-known proteins.
Membrane Blocking
Blocking buffers are essential for accurate Western blot results. They prevent antibodies from binding with the blotting membrane.
After protein blotting, the membrane is saturated with proteins but there are still empty areas. As this membrane has been specifically developed to catch and hold on to proteins – and as antibodies are also proteins – the membrane itself could affect the final result.
Without membrane blocking, a Western blot result would show the equivalent of background noise on a video recording, blurring the image. In terms of Western blot blocking buffers, this noise is a source of false-positive signals. Another term for membrane blocking is antibody probing.
Blocking buffers fill in empty spaces within the membrane. Surprisingly, skim milk powder is a commonly used buffer. Other blocking buffers are made from products such as fish gelatin, albumin, blood serum, and the non-protein polyvinylpyrrolidone (PVP).

Antibody Incubation
There are two methods of antibody incubation based on direct and indirect protein detection techniques. Direct Western blot uses a single (primary) antibody to detect a single protein. This tagged antibody binds to the target protein; the tag is a dye or enzyme that makes results visible.
The indirect Western blot method use two antibodies – a primary antibody that attaches to the target protein and a tagged secondary antibody that recognizes and marks the position of the primary antibody, making it visible.
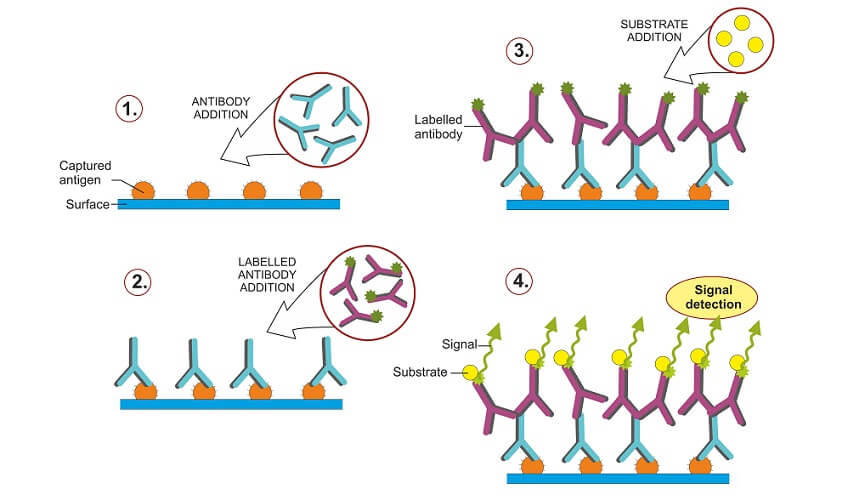
Large quantities of antibodies (immunoglobulins) are manufactured in genetically-modified animals. Monoclonal antibodies are purely man-made. They only recognize one type of antigen epitope – the part of an antigen to which immunoglobulins bind. Monoclonal antibodies are expensive but in many cases provide more accurate and consistent results than polyclonal antibodies.
Polyclonal antibodies are immunoglobulin (Ig) groups that recognize multiple epitopes. However, each antibody within this group acts like a monoclonal Ig – recognizing one epitome on one antigen. When polyclonal antibodies are used in Western blot the test is cheaper to carry out but there is a higher risk of cross-reactivity – the detection of similar but not same antigens.

Primary antibody incubation times depend on how easy it is for the Ig to attach to the target protein – its binding affinity. Average times are approximately two hours at room temperature (20°C or 68°F) or overnight at temperatures of 4°C (39.2 °F). Excessive incubation times produce more signal noise.
For indirect incubation, the membrane must be rinsed in the appropriate wash buffer numerous times before applying the secondary antibody. Secondary antibody incubation times are roughly the same as those of the primary antibody application.
Detection of the Target Protein
Detection methods vary with Western blot techniques according to which antibody tags have been used. Antibody labels can be detected with the naked eye, or with the use of X-ray film, light sensor cameras (charge-coupled device), fluorescent digital imagers, and near-infrared spectroscopy instruments.

Before detection, all primary or secondary antibodies need to be rinsed from the membrane. This prevents further reactions from occurring.
Good protein visualization is essential for correct analysis of the results. Poor protein detection is usually caused by human error or contaminated, faulty, or out-of-date solutions, materials, and equipment.
Data Analysis
Western blot data analysis today usually requires the conversion of membrane scans into digital format.
The values of protein bands are measured by comparing them to a marker of which the molecular weight is known.
Analysis needs to take several important factors into account:
- Limit of detection: some proteins are not as easily detected.
- Signal stability: other reactions, such as bacterial contamination, can cause signals to fade or even disappear over time.
- Linear dynamic range: when the limit of detection is good, the dynamic range – the proportion of protein quantity to signal intensity – is linear.
- Signal-to-noise ratio: proteins similar to target proteins are also detected – these create background noise to any target-protein signals.
Western Blot Analysis
While Western blot analysis results are commonly used to detect the presence of disease, other medical uses apply. Pharmaceutical research looks at how drugs affect other tissues, for example. Many companies make antibodies specifically for Western blot testing. However, our knowledge of specific antibodies is limited – whether these produced antibodies can detect lesser-studied antigens is a source of debate.

To produce Western blot antibodies, animals are genetically modified to not contain the gene that produces the to-be-studied protein. This means that, when exposed to it, the animal’s immune system sees the protein as a foreign invader; it begins to produce antibodies to destroy it. The genetically-modified animal is called a knockout animal. Many laboratories produce their own knockout animals rather than purchase ready-made antibodies.
Knowing how to read a Western blot is relatively simple. Bands are given a positive or negative result. Not all results are clear. Band sizes are represented by a number preceded by the letter p or followed by kDa (protein molecular weight in kilodaltons). The p refers to a specified protein density within the sample.
Western blot HIV results pick out the proteins of the human immunodeficiency virus. It measures four proteins: glycoproteins 160 and 120, and p24 and p31 antigens. Positive results must show, at minimum, one of each of the glycoprotein and antigen sets.
Western blot Lyme results for this tick-borne disease are 97% accurate but only in people who have developed Lyme arthritis or Lyme carditis. The detected bacterial proteins are from blood-borne Borrelia burgdorferi.

The University of Washington’s Western blot herpes detection test used to be the gold standard in testing but has since been shown to produce overly-high false-positive results. Secondary testing is often required, making this an expensive screening method.
Western Blot vs ELISA
ELISA stands for enzyme-linked immunosorbent assay and works in the opposite way as the Western blot. Instead of detecting antigens by way of antibodies, ELISA looks at the presence of antibodies by binding them to known antigens.
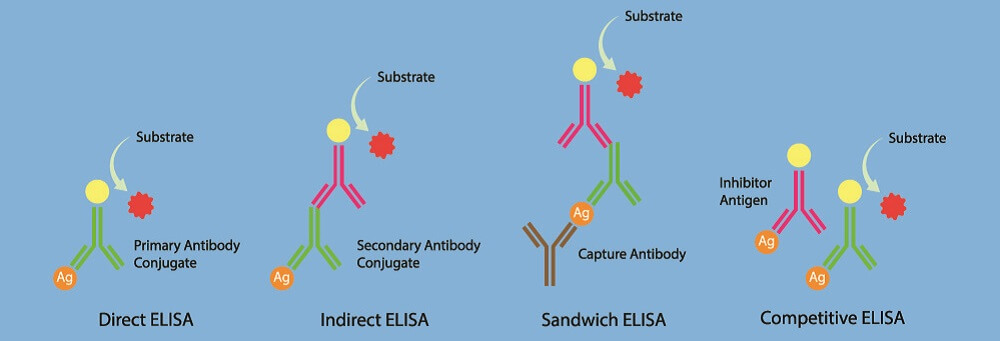
If a person has been infected with a virus in the past but is no longer ill, Western blot will not pick up these viral antigens as there are no viruses left in the body. However, ELISA can detect the antibodies that were produced after an earlier infection.
Researchers are now looking at the use of N195 protein as an antigen for early Western blot detection of an active SARS coronavirus infection. ELISA, on the other hand, is already available in kit form to detect the presence of two types of COVID-associated immunoglobulins. If IgM is detected, this indicates early-onset disease. IgG detection without abnormal IgM levels would indicate a past infection.
