Glycogen Definition
Glycogen is a large, branched polysaccharide that is the main storage form of glucose in animals and humans. Glycogen is as an important energy reservoir; when energy is required by the body, glycogen in broken down to glucose, which then enters the glycolytic or pentose phosphate pathway or is released into the bloodstream. Glycogen is also an important form of glucose storage in fungi and bacteria.
Glycogen Structure
Glycogen is a branched polymer of glucose. Glucose residues are linked linearly by α-1,4 glycosidic bonds, and approximately every ten residues a chain of glucose residues branches off via α-1,6 glycosidic linkages. The α-glycosidic bonds give rise to a helical polymer structure. Glycogen is hydrated with three to four parts water and forms granules in the cytoplasm that are 10-40nm in diameter. The protein glycogenin, which is involved in glycogen synthesis, is located at the core of each glycogen granule. Glycogen is an analogue of starch, which is the main form of glucose storage in most plants, but starch has fewer branches and is less compact than glycogen.
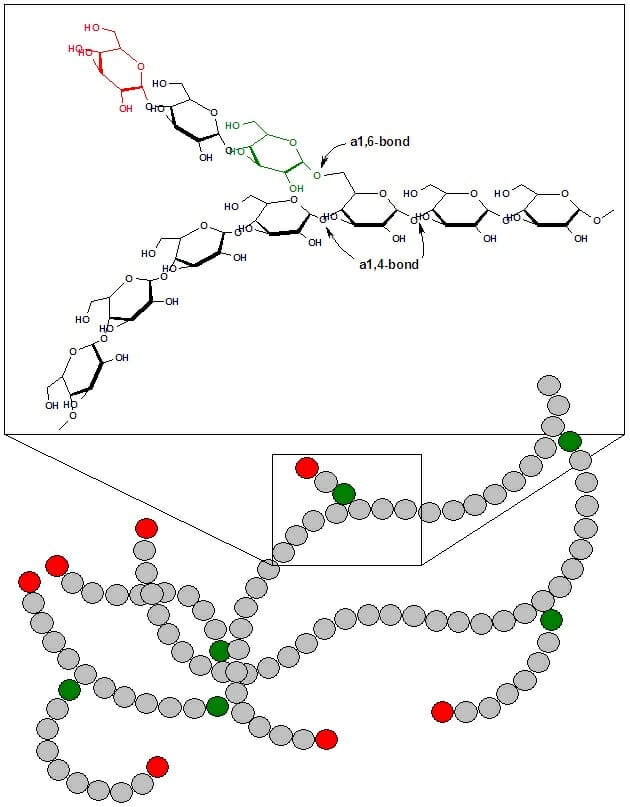
This figures shows the structure of glycogen. Green circles represent α-1,6 linkages at branch points, and red circles represent the nonreducing ends of the chain.
Glycogen Function
In animals and humans, glycogen is found mainly in muscle and liver cells. Glycogen is synthesized from glucose when blood glucose levels are high, and serves as a ready source of glucose for tissues throughout the body when blood glucose levels decline.
Liver Cells
Glycogen makes up 6-10% of the liver by weight. When food is ingested, blood glucose levels rise, and insulin released from the pancreas promotes the uptake of glucose into liver cells. Insulin also activates enzymes involved in glycogen synthesis, such as glycogen synthase. While glucose and insulin levels are sufficiently high, glycogen chains are elongated by the addition of glucose molecules, a process termed glyconeogenesis. As glucose and insulin levels decrease, glycogen synthesis ceases. When blood glucose levels fall below a certain level, glucagon released from the pancreas signals to liver cells to break down glycogen. Glycogen is broken down via glycogenolysis into glucose-1-phosphate, which is converted to glucose and released into the bloodstream. Thus, glycogen serves as the main buffer of blood glucose levels by storing glucose when it levels are high and releasing glucose when levels are low. Glycogen breakdown in the liver is critical for supplying glucose to meet the body’s energetic needs. In addition to glucagon, cortisol, epinephrine, and norepinephrine also stimulate glycogen breakdown.
Muscle Cells
In contrast to liver cells, glycogen only accounts for 1-2% of muscle by weight. However, given the greater mass of muscle in the body, the total amount of glycogen stored in muscle is greater than that stored in liver. Muscle also differs from liver in that the glycogen in muscle only provides glucose to the muscle cell itself. Muscle cells do not express the enzyme glucose-6-phosphatase, which is required to release glucose into the bloodstream. The glucose-1-phosphate produced from glycogen breakdown in muscle fibers is converted to glucose-6-phosphate and provides energy to the muscle during a bout of exercise or in response to stress, as in the fight-or-flight response.
Other Tissues
In addition to liver and muscle, glycogen in found in smaller amounts in other tissues, including red blood cells, white blood cells, kidney cells, and some glial cells. Additionally, glycogen is used to store glucose in the uterus to provide for the energetic needs of the embryo.
Fungi and Bacteria
Microorganisms possess mechanisms for storing energy to cope in the event of limited environmental resources, and glycogen represents a main energy storage form. Nutrient limitation (low levels of carbon, phosphorus, nitrogen, or sulfur) can stimulate glycogen formation in yeast, while bacteria synthesize glycogen in response to readily available carbon energy sources with limitation of other nutrients. Bacterial growth and yeast sporulation have also been associated with glycogen accumulation.
Glycogen Metabolism
Glycogen homeostasis is a highly regulated process that allows the body to store or release glucose depending on its energetic needs. The basic steps in glucose metabolism are glycogenesis, or glycogen synthesis, and glycogenolysis, or glycogen breakdown.
Glycogenesis
Glycogen synthesis requires energy, which is supplied by uridine triphosphate (UTP). Hexokinases or glucokinase first phosphorylate free glucose to form glucose-6-phosphate, which is converted to glucose-1-phosphate by phosphoglucomutase. UTP-glucose-1-phosphate uridylyltransferase then catalyzes the activation of glucose, in which UTP and glucose-1-phosphate react to form UDP-glucose. In de novo glycogen synthesis, the protein glycogenin catalyzes the attachment of UDP-glucose to itself. Glycogenin is a homodimer containing a tyrosine residue in each subunit that serves as an anchor or attachment point for glucose. Additional glucose molecules are subsequently added to the reducing end of the previous glucose molecule to form a chain of approximately eight glucose molecules. Glycogen synthase then extends the chain by adding glucose via α-1,4 glycosidic linkages.
Branching is catalyzed by amylo-(1,4 to 1,6)-transglucosidase, also called the glycogen branching enzyme. The glycogen branching enzyme transfers a fragment of six to seven glucose molecules from the end of a chain to the C6 of a glucose molecule located further inside the glycogen molecule, forming α-1,6 glycosidic linkages.
Glycogenolysis
Glucose is removed from glycogen by glycogen phosphorylase, which phosphorolytically removes one molecule of glucose from the nonreducing end, yielding glucose-1-phosphate. The glucose-1-phosphate generated by glycogen breakdown is converted to glucose-6-phosphate, a process that requires the enzyme phosphoglucomutase. Phosphoglucomutase transfers a phosphate group from a phosphorylated serine residue within the active site to C6 of glucose-1-phosphate, producing glucose-1,6-bisphosphate. The glucose C1 phosphate is then attached to the active site serine within phosphoglucomutase, and glucose-6-phosphate is released.
Glycogen phosphorylase is not able to cleave glucose from branch points; debranching requires amylo-1,6-glucosidase, 4-α-glucanotransferase, or glycogen debranching enzyme (GDE), which has glucotransferase and glucosidase activities. About four residues from a branch point, glycogen phosphorylase is unable to remove glucose residues. GDE cleaves the final three residues of a branch and attaches them to C4 of a glucose molecule at the end of a different branch, then removes the final α-1,6-linked glucose residue from the branch point. GDE does not remove the α-1,6-linked glucose from the branch point phosphorylytically, meaning that free glucose is released. This free glucose could in theory be released from muscle into the bloodstream without the action of glucose-6-phosphatase; however this free glucose is rapidly phosphorylated by hexokinase, preventing it from entering the bloodstream.
The glucose-6-phosphate resulting from glycogen breakdown may be converted to glucose by the action of glucose-6-phosphatase and released into the bloodstream. This occurs in liver, intestine, and kidney, but not in muscle, where this enzyme is absent. In muscle, glucose-6-phosphate enters the glycolytic pathway and provides energy to the cell. Glucose-6-phosphate may also enter the pentose phosphate pathway, resulting in the production of NADPH and five carbon sugars.
Exercise and Glycogen Depletion
In endurance exercise, athletes may undergo glycogen depletion, in which most of the glycogen is depleted from the muscle. This can result in severe fatigue and difficulty moving. Glycogen depletion can be mitigated by continuously consuming carbohydrates with a high glycemic index (high rate of conversion to blood glucose) during exercise, which will replace some of the glucose using during exercise. Specialized exercise regimens may also be employed that condition muscle to utilize fatty acids as an energy source at a greater rate, thus breaking down less glycogen. Athletes may also use carbohydrate loading, the consumption of large amounts of carbohydrates, to increase the capacity for glycogen storage.
Examples of Glycogen Storage Diseases
There are two main categories of glycogen storage diseases: those resulting from defective glycogen homeostasis in the liver and those resulting from defective glycogen homeostasis in the muscle. Diseases resulting from defective liver glycogen storage generally cause hepatomegaly (enlarged liver), hypoglycemia, and cirrhosis (liver scarring). Diseases resulting from defective muscle glycogen storage generally cause myopathies and metabolic impairment. Examples of glycogen storage diseases include Pompe Disease, McArdle Disease, and Andersen Disease.
Pompe Disease
Pompe disease is caused by mutations in the GAA gene, which encodes lysosomal acid α-glucosidase, also referred to as acid maltase, and affects skeletal and cardiac muscle. Acid maltase is involved in glycogen breakdown, and disease-causing mutations result in the detrimental buildup of glycogen in the cell. There are three types of Pompe Disease: the adult form, juvenile form, and infantile form, which are progressively more severe. The infantile form leads to death by one to two years of age if left untreated.
McArdle Disease
McArdle Disease is caused by mutations in the PYGM gene, which encodes myophosphorylase, the glycogen phosphorylase isoform present in muscle. Symptoms are often observed in children, but the disease may not be diagnosed until adulthood. Symptoms include muscle pain and fatigue, and the disease may be life-threatening if not properly treated.
Andersen Disease
Andersen Disease is caused by a mutation in the GBE1 gene, which encodes glycogen branching enzyme, and affects muscle and liver. Symptoms are usually observed at a few months of age, and include stunted growth, liver enlargement, and cirrhosis. Complications of the disease can be life-threatening.
Quiz
1. Which best describes the function of glycogen?
A. Provides structural support to muscle cells
B. A transcription factor that regulates cell differentiation
C. Stores glucose in plants
D. Buffers blood glucose levels and serves as a readily mobilized energy source
2. What is the main hormone that stimulates glycogen breakdown?
A. Glucagon
B. Thyroid
C. Insulin
D. Estrogen
3. What are the possible fates of the glucose-1-phosphate produced by glycogenolysis?
A. Conversion to glucose-6-phosphate followed by entrance into the glycolytic pathway
B. Conversion to glucose-6-phosphate followed by entrance into the pentose phosphate pathway
C. Conversion to glucose followed by release into the bloodstream
D. All of the above
References
- Eicke, S., Seung, D., Egli, B., Devers, E.A., and Streb, S. (2017) “Increasing the carbohydrate storage capacity of plants by engineering a glycogen-like polymer pool in the cytosol.” Metabolic Engineering. 40:23-32.
- Hargreaves, M. and Richter, E.A. (1988) “Regulation of skeletal muscle glycogenolysis during exercise.” Canadian Journal of Sport Sciences. 13(4):197-203.
- Ivy, J.L. (1991). “Muscle glycogen synthesis before and after exercise.” Sports Medicine. 11(1):6-19.