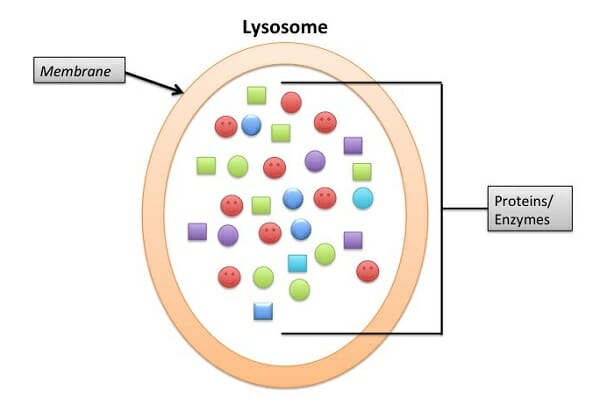
Lysosome Definition
Lysosomes are specialized vesicles within cells that digest large molecules through the use of hydrolytic enzymes. Vesicles are small spheres of fluid surrounded by a lipid bilayer membrane, and they have roles in transporting molecules within the cell. Lysosomes are only found in animal cells; a human cell contains around 300 of them. Not only do they digest large molecules, they are also responsible for breaking down and getting rid of waste products of the cell. Lysosomes contain over 60 different enzymes that allow them to carry out these processes.
Functions of the Lysosome
Lysosomes digest many complex molecules such as carbohydrates, lipids, proteins, and nucleic acids, which the cell then recycles for other uses. The pH of lysosomes is acidic (around pH 5) because their hydrolytic enzymes function best at this pH instead of at the neutral pH of the rest of the cell. Hydrolytic enzymes specifically break down large molecules through hydrolysis. During the process of hydrolysis, a molecule of water is added to a substance, causing it to cleave. Like the digestive system of the human body, which breaks down food using enzymes, the lysosome can be thought of as the “digestive system” of the cell because it breaks down molecules using enzymes.
Lysosomes digest several different kinds of molecules. They can digest food molecules that enter the cell into smaller pieces if an endocytic vesicle (a vesicle that brings particles into the cell) fuses with them. They can also perform autophagy, which is the destruction of improperly functioning organelles. In addition, lysosomes have a role in phagocytosis, which is when a cell engulfs a molecule in order to break it down; it is also known as “cell eating”. For example, the white blood cells called phagocytes ingest invading bacteria in order to break it down and destroy it, and the bacteria is enclosed by a vesicle that lysosomes fuse with. These lysosomes then break down the bacteria.
Lysosome Structure
Lysosomes are generally very small, ranging in size from 0.1-0.5 µm, though they can reach up to 1.2 µm. They have a simple structure; they are spheres made up of a lipid bilayer that encloses fluid that contains a variety of hydrolytic enzymes. The lipids that make up the bilayer are phospholipids, which are molecules that have hydrophilic phosphate group heads, a glycerol molecule, and hydrophobic fatty acid tails. Due to these differences in properties, phospholipids naturally form double-layered membranes when placed in a solution containing water. The phosphate group heads move to the outside of the layer, while the fatty acid tails move to the inside of the layer to be away from water. Phospholipids make up many other membranes in the cell, such as the cell membrane which surrounds the entire cell, the nuclear membrane (or nuclear envelope) that surrounds the nucleus, the Golgi apparatus, and the endoplasmic reticulum.
Lysosomes are formed by budding off of the Golgi apparatus, and the hydrolytic enzymes within them are formed in the endoplasmic reticulum. The enzymes are tagged with the molecule mannose-6-phosphate, transported to the Golgi apparatus in vesicles, and then packaged into the lysosomes.
There are many different types of enzymes in lysosomes including proteases, amylases, nucleases, lipases, and acid phosphatases, among many others. Enzymes are usually named for the molecules that they break down; for example, proteases break down proteins, and nucleases break down nucleic acids. Amylases break down starches into sugars.
The following images are a simplified structure of the lysosome and a more detailed depiction of the phospholipid bilayer structure.
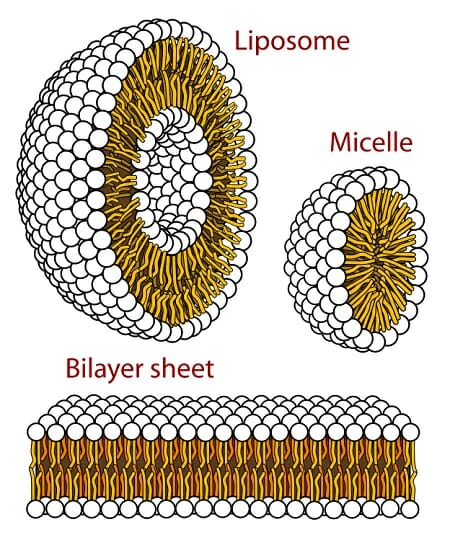
Liposomes, not to be confused with lysosomes, are artificially created vesicles that, like all vesicles including lysosomes, have phospholipid bilayers. They are sometimes used to deliver nutrients and pharmaceutical drugs.
Lysosomal Storage Diseases
Some inherited metabolic disorders can cause defects in the proper functioning of lysosomes. These disorders are called lysosomal storage diseases, or LSDs. There are around 50 different LSDs. Each type of LSD is rare, occurring in less than 1 in 100,000 births; however, as a group, LSDs occur in 1 in 5,000-10,000. LSDs usually occur when a person is deficient in one enzyme that breaks down large molecules like proteins or lipids. Because the enzyme is lacking, the large molecules cannot be broken down, and they eventually build up within the cell and kill it.
Most LSDs are inherited in an autosomal recessive pattern. This means that it can be masked by a copy of an allele without the mutation (a dominant allele) and is caused by a mutation on one of the autosomal chromosomes, which are all chromosomes except the sex chromosomes X and Y. Tay-Sachs disease is an example of a well-known LSD that is recessively inherited. Due to insufficient function of the enzyme hexosaminidase A, glycolipids build up in the brain and interfere with normal functioning. This causes nerve cells to break down, and physical and mental functioning to decline. There is no cure, and death usually occurs by age four.
A few LSDs are X-linked; they occur because of a mutation on the X chromosome. One such LSD is Fabry disease. Fabry disease is rare, occurring in 1 in 40,000-120,000 live births. People with Fabry disease are deficient in the enzyme alpha galactosidase A, which causes the glycolipid globotriaosylceramide to build up within the body. Symptoms include fatigue, burning pain in the extremities or full body pain, tinnitus, nausea, cardiac and kidney complications, and papules on the skin called angiokeratomas. The mutation that causes Fabry disease is located on the X-chromosome, but females with only one copy of the mutated gene also show symptoms. Since men only have one X chromosome, their symptoms tend to be more severe. Life expectancy for those with this disease in the United States is 58.2 for males and 75.4 for females.
Related Biology Terms
- Vesicle – A small sphere of lipid bilayer in the cell that can transport molecules.
- Lysosomal storage diseases (LSDs) – A group of about 50 genetic disorders involving abnormal lysosomal function.
- Autophagy – The degradation of unnecessary or improperly functioning components within a cell.
- Hydrolytic enzyme – A molecule that speeds up a chemical reaction involving hydrolysis.
Quiz
1. How can lysosomal storage disorders be inherited?
A. Autosomal recessive
B. X-linked recessive
C. Autosomal dominant
D. Choices A and B
2. How many different hydrolytic enzymes do lysosomes contain?
A. 20+
B. 30+
C. 50+
D. 60+
3. What is the mechanism by which most LSDs occur?
A. The lysosomes are too small to contain the large molecules that they normally can break down.
B. Enzymes are not packaged into the lysosomes in the Golgi apparatus correctly.
C. Deficiency in one hydrolytic enzyme in lysosomes leads to a buildup of large molecules, eventually killing the cell.
D. The phospholipid bilayer of lysosomes does not form correctly, so the lysosomes cannot contain the necessary enzymes.