Mitochondrion Definition
The mitochondrion (plural mitochondria) is a membrane-bound organelle found in the cytoplasm of eukaryotic cells. It is the power house of the cell; it is responsible for cellular respiration and production of (most) ATP in the cell. Each cell can have from one to thousands of mitochondria. Mitochondria also contain extranuclear DNA that encodes a number of rRNAs, tRNAs, and proteins.
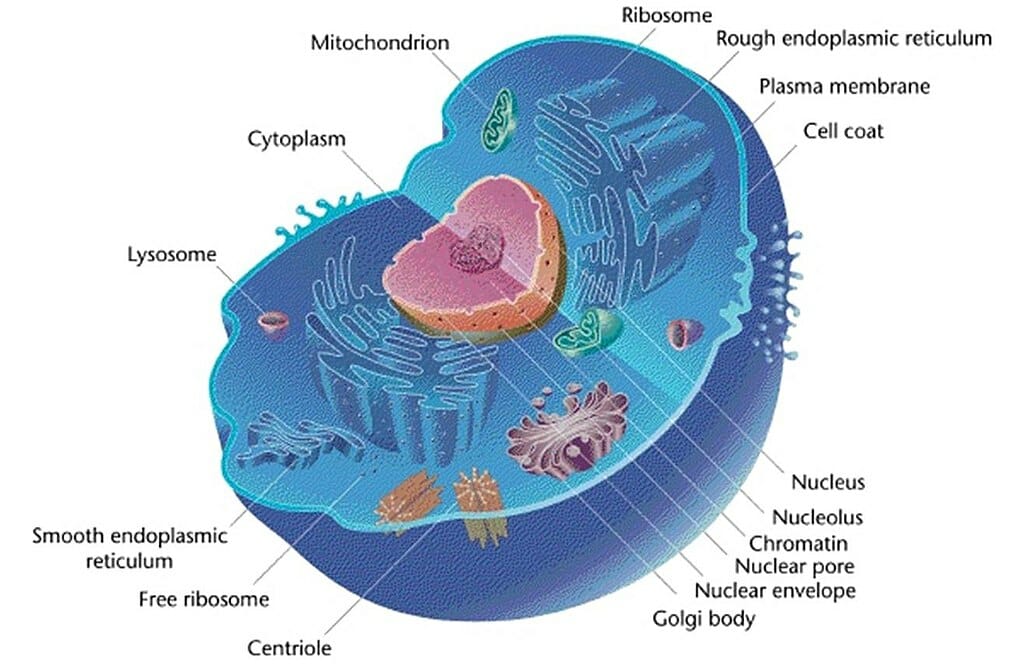
The figure depicts the general structure of a typical animal cell. The organelles are labelled.
Mitochondrion Origin
The current theory as to the origin of eukaryotic cells is endosymbiosis. It is believed that mitochondria (and chloroplasts) began as prokaryotic organisms that were living within larger cells. It is likely that these prokaryotic organisms were engulfed by the larger cells, either as food or parasites. At some point the relationship became mutually beneficial and the mitochondria and chloroplasts became a permanent feature in the cells. They were enclosed in membranes and formed cellular machinery.
Mitochondrion Structure
Mitochondria are small membrane-bound organelles that are usually about 1 – 10 microns in length. They can be spherical or rod-shaped. The mitochondrion is enclosed by two membranes that separate it from the cytosol and the rest of the cell components. The membranes are lipid bilayers with proteins embedded within the layers. The inner membrane is folded to form cristae; this increases the surface area of the membrane and maximizes cellular respiration output. The region between the two membranes is the intermembrane space. Inside the inner membrane is the mitochondrial matrix, and within the matrix there are ribosomes, other enzymes, and mitochondrial DNA. The mitochondrion is able to reproduce and synthesize proteins independently. It contains the enzymes necessary for transcription, as well as the transfer RNAs and ribosomes required for translation and protein formation.
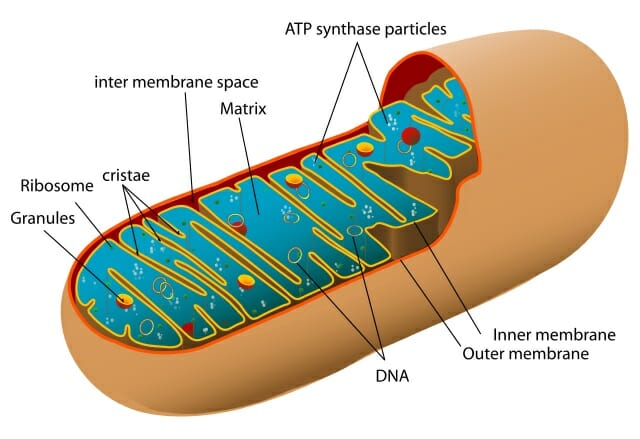
The figure shows a cut-out of an animal mitochondrion. The major components are labelled.
Mitochondrial DNA
Mitochondrial DNA (mtDNA) is typically a small circular double-stranded DNA molecule that encodes a number of proteins and RNA involved primarily in cellular respiration and cell reproduction. In some protists and fungi, mtDNA can be linear. Mitochondrial DNA is well conserved within taxa. For example, many birds or mammals have the same gene order. Animal mitochondrial DNA encodes two ribosomal RNAs, 22 transfer RNAs, and 13 protein coding genes (subunits of NADH, ATPase, and cytochromes). It also consists of the non-coding control region, or D-loop, which is involved in the regulation of DNA replication.
Unlike nuclear DNA, which is passed on from both parents, mitochondrial DNA is generally uniparentally inherited (with some notable exceptions). In animals mtDNA is passed on maternally through the egg, except in bivalve molluscs where biparental inheritance is found. In plants mtDNA may be passed on maternally, paternally, or biparentally. There is also evidence for paternal leakage of mtDNA, where the offspring inherits most of their mtDNA from their mother but also receives a small amount from their father.
Mutations in mitochondrial DNA can result in a number of human genetic diseases, particularly those that involve energy consumption in the muscular and nervous systems. Examples include diabetes, heart disease, myoclonic epilepsy, Kearns-Sayre neuromuscular syndrome, and Alzheimer’s. It has also been implicated in degenerative diseases and aging.
Compared to nuclear coding genes, animal mitochondrial DNA evolves about 10 times more quickly, allowing changes to be seen in a relatively short time frame. It also mutates in a relatively clock-like fashion (with some exceptions). For this reason mitochondrial DNA is commonly used to study evolutionary relationships and population genetics in animals; it was the driving force behind the “Out-of-Africa” hypothesis of human evolution, as well as the evolutionary relationship between humans and apes. Plant mtDNA evolves fairly slowly, and is less commonly used in evolutionary studies.
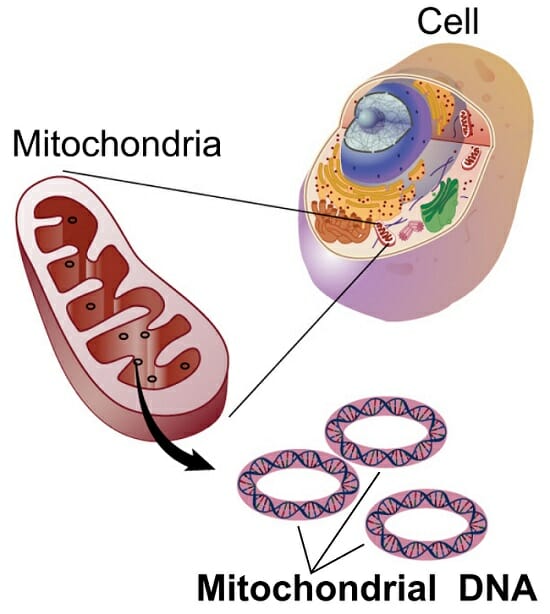
The figure shows the small circular DNA molecules within the organelles.
Mitochondrion Function
Mitochondria are involved in breaking down sugars and fats into energy through aerobic respiration (cellular respiration). This metabolic process creates ATP, the energy source of a cell, through a series of steps that require oxygen. Cellular respiration involves three main stages.
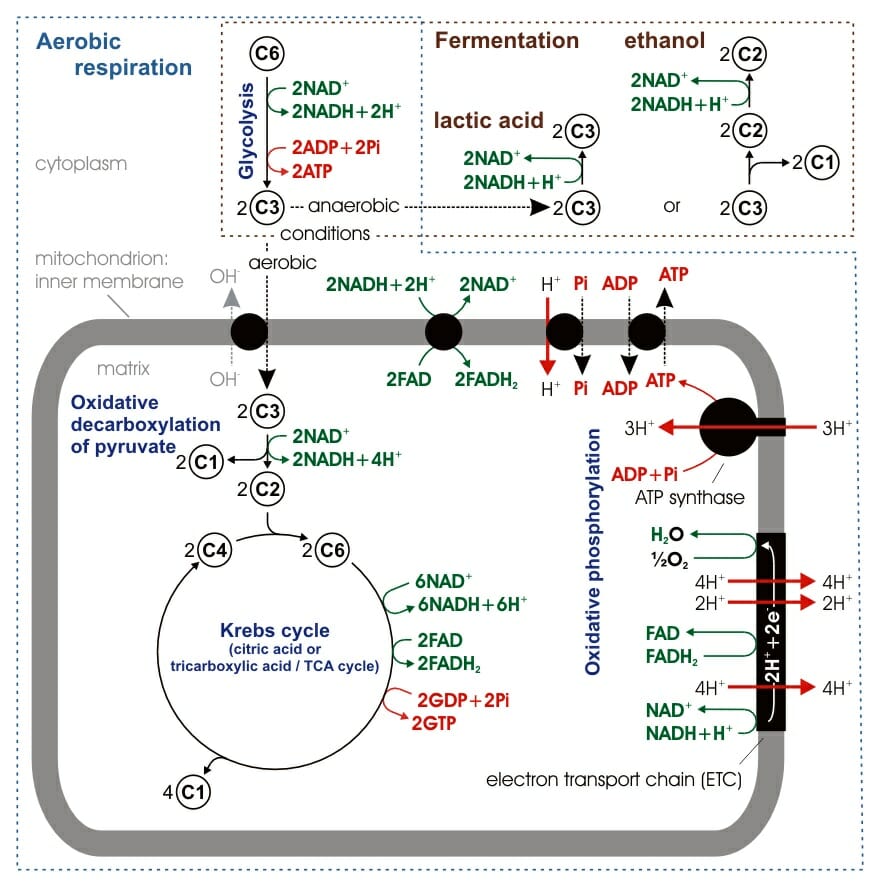
The figure shows an overview of cellular respiration. Glycolysis takes place in the cytosol while the Krebs cycle and oxidative phosphorylation occur in the mitochondria.
Glycolysis
Glycolysis occurs in the cytosol, splitting glucose into two smaller sugars which are then oxidized to form pyruvate. Glycolysis can be either anaerobic or aerobic, and as such is not technically part of cellular respiration, although it is often included. It produces a small amount of ATP.
During glycolysis the starting glucose molecule is phosphorylated (using one ATP molecule), forming glucose-6-phosphate, which then rearranges to its isomer fructose-6-phosphate. The molecule is again phosphorylated (using a second ATP molecule), this time forming fructose-1,6-bisphosphate. Fructose-1,6-bisphosphate is then split into two 3-carbon sugars which are converted to pyruvate molecules through a redox reaction, which produces two NADH molecules, and substrate-level phosphorylation, which releases four molecules of ATP. Glycolysis produces a net two ATP molecules.
Citric Acid Cycle
In the presence of oxygen, the pyruvate molecules produced in glycolysis enter the mitochondrion. The citric acid cycle, or Krebs cycle, occurs in the mitochondrial matrix. This process breaks down pyruvate into carbon dioxide in an oxidation reaction. The citric acid cycle results in the formation of NADH (from NAD+) which transports electrons to the final stage of cellular respiration. The citric acid cycle produces two ATP molecules.
Pyruvate enters the mitochondrion and is converted into acetyl coenzyme A. This conversion is catalyzed by enzymes, produces NADH, and releases CO2. The acetyl group then enters the citric acid cycle, a series of eight enzyme-catalyzed steps that begins with citrate and ends in oxaloacetate. The addition of the acetyl group to oxaloacetate forms citrate and the cycle repeats. The breakdown of citrate into oxaloacetate releases a further two CO2 molecules and one molecule of ATP (through substrate-level phosphorylation). The majority of the energy is in the reduced coenzymes NADH and FADH2. These molecules are then transported to the electron transport chain.
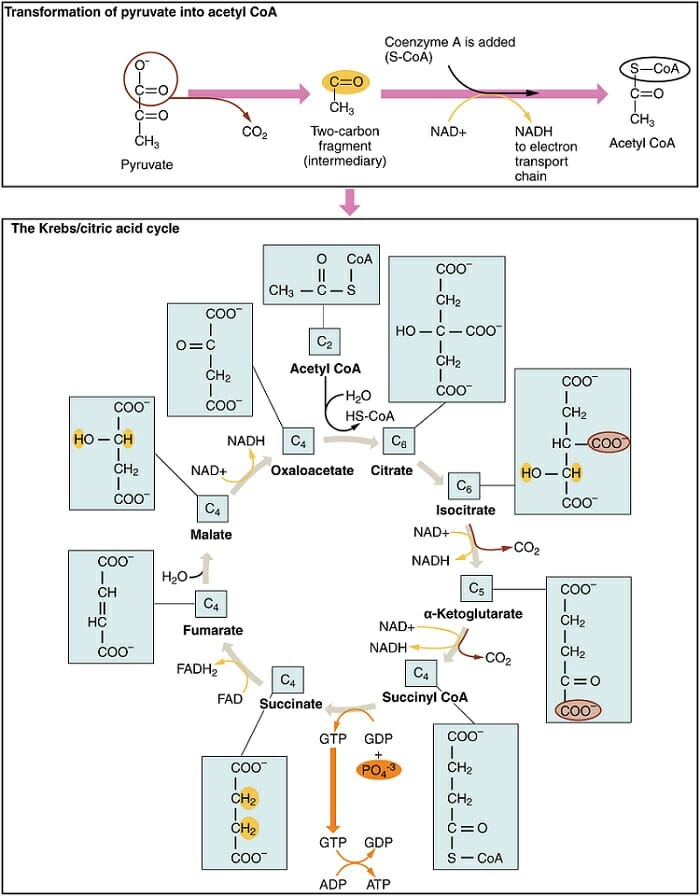
The figure shows the conversion of pyruvate into acetyl coenzyme A and its progression through the citric acid cycle.
Oxidative Phosphorylation
Oxidative phosphorylation consists of two parts: the electron transport chain and chemiosmosis. It is this final stage that produces the bulk of the ATP in the respiration process. The electron transport chain uses the electrons carried forward from the previous two steps (as NADH and FADH2) to form water molecules through combination with oxygen and hydrogen ions. Oxidative phosphorylation occurs in the inner membrane of the mitochondrion.
The electron transport chain is made up of five multi-protein complexes (I to IV) that are repeated hundreds to thousands of times in the cristae of the inner membrane. The complexes are made up of electron carriers that transport the electrons released from NADH and FADH2 through a series of redox reactions. Many of the proteins found in the electron transport chain are cytochromes, proteins that are encoded for in part by mitochondrial DNA. As the electrons move along the chain they are passed to increasingly more electronegative molecules. The final step is the transfer of the electron to an oxygen atom which combines with two hydrogen ions to form a water molecule. The electron transport chain itself does not produce ATP.
ATP is produced via chemiosmosis, a process that also occurs in the inner membrane of the mitochondrion. Chemiosmosis involves the transmembrane protein ATP synthase which produces ATP from ADP and inorganic phosphate. ATP synthase uses the concentration gradient of hydrogen ions to drive the formation of ATP. As the electrons move through the electron transport chain, hydrogen ions are pushed out into the intermembrane space, producing a higher concentration of H+ outside the membrane. The consumption of H+ through incorporation into water molecules further increases the concentration gradient. The hydrogen ions then try to re-enter the mitochondrial matrix to equalize the concentrations; the only place they can cross the membrane is through the ATP synthase. The flow of H+ through the enzyme results in conformational changes that provide catalytic active sites for ADP and inorganic phosphate. When these two molecules bind to the ATP synthase they are connected and catalyzed to form ATP.
Oxidative phosphorylation produces between 32 and 34 ATP molecules from each initial glucose molecule, accounting for ~89% of the energy produced in cellular respiration.
Quiz
1. Which step of cellular respiration produces the most ATP?
A. Krebs cycle
B. Glycolysis
C. Citric acid cycle
D. Chemiosmosis
2. Where does oxidative phosphorylation occur?
A. Mitochondrial matrix
B. Outer membrane
C. Inner membrane
D. Intermembrane space
3. What organisms do not contain mitochondria?
A. Plants
B. Animals
C. Bacteria
D. Fungi
References
- Boore, J. L. (1999). Animal mitochondrial genomes. Nucleic Acids Research, 27, 1767-1780.
- Brown, W. M., George, M., & Wilson, A. C. (1979). Rapid evolution of animal mitochondrial DNA. Proceedings of the National Academy of Sciences USA, 76, 1967-1971.
- Campbell, N. A., & Reece, J. B. (2005).Biology, 7th. ed. Chs. 6, 9, and 26. San Francisco, CA: Benjamin Cummings. ISBN: 0-8053-7171-0.
- Cann, R. L., Stoneking, M., & Wilson, A. C. (1987). Mitochondrial DNA and human evolution. [10.1038/325031a0]. Nature, 325, 31-36.
- Madigan, M. T., & Martinko, J. M. (2006).Brock biology of microorganisms, 11th. ed. Chs. 7 and 14. Upper Saddle River, NJ: Pearson Prentice Hall. ISBN: 0-13-144329-1.
- Wallace, D. C. (1999). Mitochondrial diseases in man and mouse. Science, 283, 1482-1488.