DNA Sequencing Definition
DNA sequencing is the process of determining the sequence of nucleotides within a DNA molecule. Every organism’s DNA consists of a unique sequence of nucleotides. Determining the sequence can help scientists compare DNA between organisms, which can help show how the organisms are related.
DNA Sequencing Overview
This means that by sequencing a stretch of DNA, it will be possible to know the order in which the four nucleotide bases – adenine, guanine, cytosine, and thymine – occur within that nucleic acid molecule.
The necessity of DNA sequencing was first made obvious by Francis Crick’s theory that the sequence of nucleotides within a DNA molecule directly influenced the amino acid sequences of proteins. At the time, the belief was that a completely sequenced genome would lead to a quantum leap in understanding the biochemistry of cells and organisms.
Modern DNA sequencing consists of high-throughput methods which allow entire DNA sequences to be discovered in a matter of hours. This technology has allowed many companies to start offering at-home DNA testing. Many of the “results” found by these tests are simply correlations found between a genetic variant and a certain condition. However, technology has also allowed scientists to test the DNA of many organisms to better understand evolutionary relationships.
DNA Sequencing Example
Though DNA sequencing used to take years, it can now be done in hours. Further, the first full sequence of human DNA took around 3 billion dollars. Now, certain companies will sequence your entire genome for less than $1,000. The most advanced tests will analyze every nucleotide within your genome. However, many companies now offer single-nucleotide polymorphism tests.
These tests focus on individual nucleotides within genes that can signify certain genetic variants. These SNPs, as they are known, have been correlated to certain conditions and can help predict how your genes may influence your life. Some SNPs are related to various diseases, while others are related to your metabolism and how your body processes nutrients. Thousands of different correlations have been found, and DNA sequencing can be used to figure out how your genome affects your life.
DNA Sequencing Methods
There are two main types of DNA sequencing. The older, classical chain termination method is also called the Sanger method. Newer methods that can process a large number of DNA molecules quickly are collectively called High-Throughput Sequencing (HTS) techniques or Next-Generation Sequencing (NGS) methods.
Sanger Sequencing
The Sanger method relies on a primer that binds to a denatured DNA molecule and initiates the synthesis of a single-stranded polynucleotide in the presence of a DNA polymerase enzyme, using the denatured DNA as a template. In most circumstances, the enzyme catalyzes the addition of a nucleotide. A covalent bond, therefore, forms between the 3′ carbon atom of the deoxyribose sugar molecule in one nucleotide and the 5′ carbon atom of the next. This image below shows how this bond is formed.
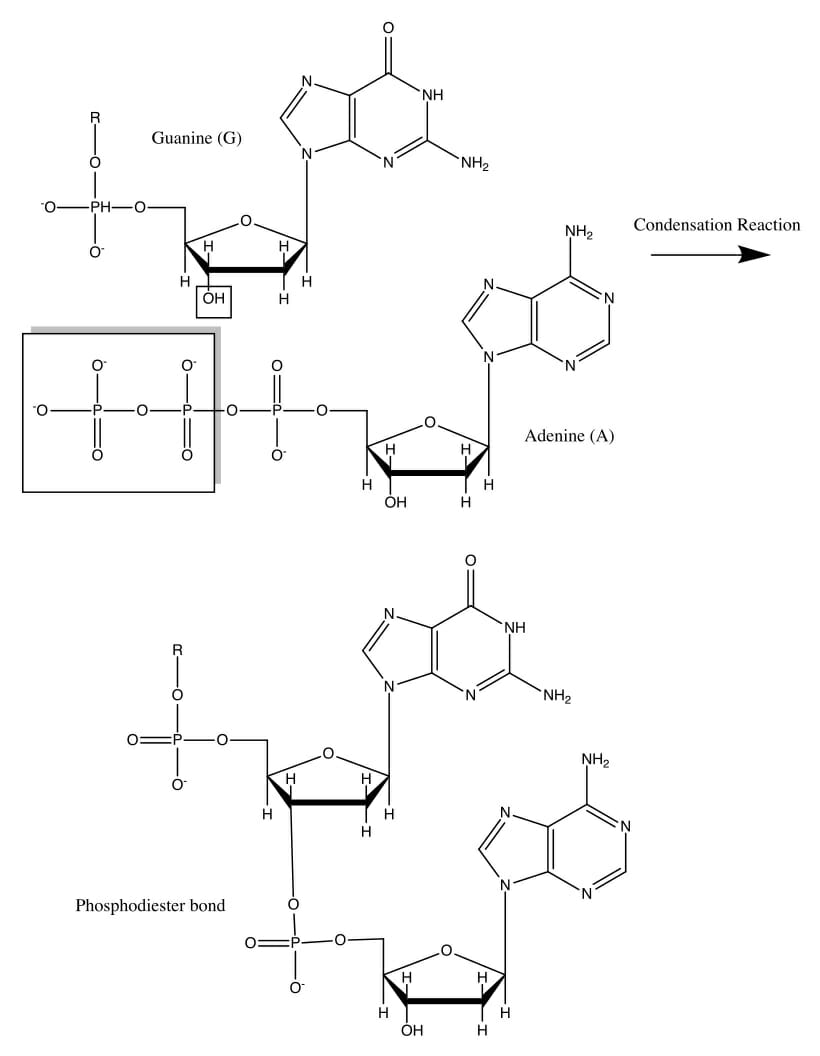
A sequencing reaction mixture, however, would have a small proportion of modified nucleotides that cannot form this covalent bond due to the absence of a reactive hydroxyl group, giving rise to the term ‘dideoxyribonucleotides’, i.e., they do not have a 2’ or 3’ oxygen atom when compared to the corresponding ribonucleotide. This would terminate the DNA polymerization reaction prematurely. At the end of multiple rounds of such polymerizations, a mixture of molecules of varying lengths would be created.
In the earliest attempts at using the Sanger method, the DNA molecule was first amplified using a labeled primer and then split into four test tubes, each having only one type of ddNTP. That is, each reaction mixture would have only one type of modified nucleotide that could cause chain termination. After the four reactions were completed, the mixture of DNA molecules created by chain termination would undergo electrophoresis on a polyacrylamide gel, and get separated according to their length.
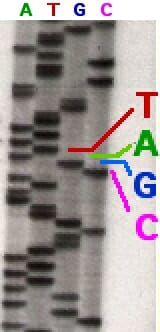
In the image above, a sequencing reaction with ddATP was electrophoresed through the first column. Each line represents a DNA molecule of a particular length, the result of a polymerization reaction that was terminated by the addition of a ddATP nucleotide. The second, third and fourth columns contained ddTTP, ddGTP, and ddCTP respectively.
With time, this method was modified so that each ddNTP had a different fluorescent label. The primer was no longer the source of the radiolabel or fluorescent tag. Also known as dye-terminator sequencing, this method used four dyes with non-overlapping emission spectra, one for each ddNTP.
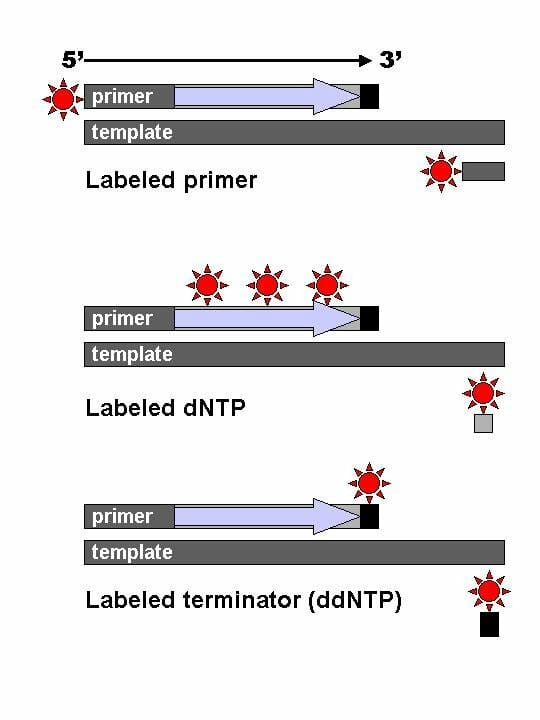
The image shows the difference between labeled primers, labeled dNTPs and dyed terminator NTPs.
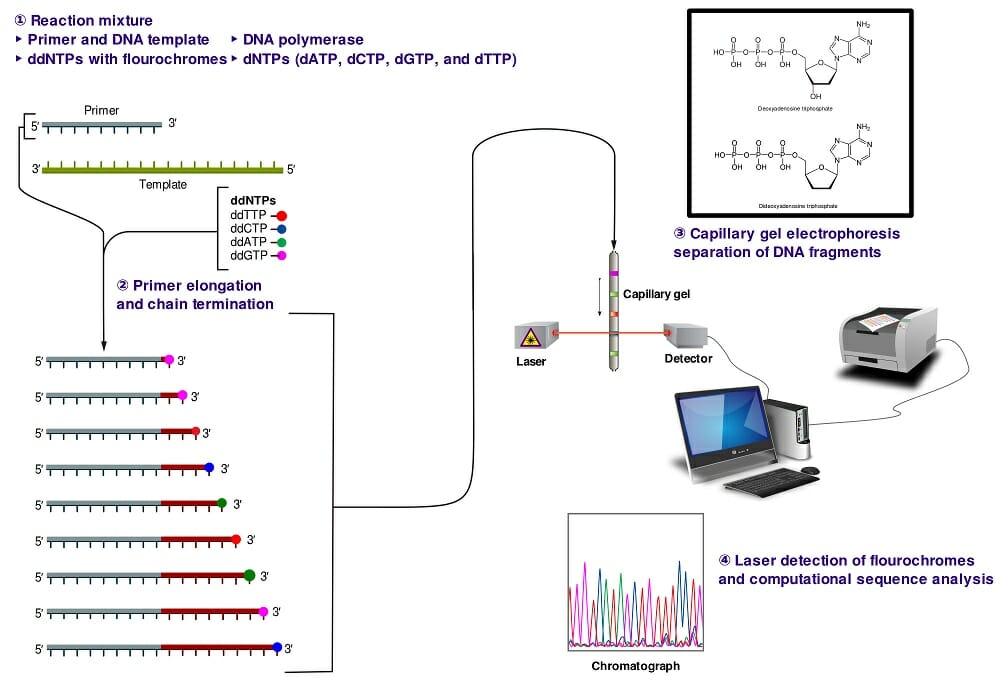
The image above shows a schematic representation of dye-terminator sequencing. There is a single reaction mixture carrying all the elements needed for DNA elongation. The reaction mixture also contains small concentrations of four ddNTPs, each with a different fluorescent tag. The completed reaction is run on a capillary gel. The results are obtained through an analysis of the emission spectra from each DNA band on the gel. A software program then analyzes the spectra and presents the sequence of the DNA molecule.
High Throughput Sequencing
Sanger sequencing continues to be useful for determining the sequences of relatively long stretches of DNA, especially at low volumes. However, it can become expensive and laborious when a large number of molecules need to be sequenced quickly. Ironically, though the traditional dye-terminator method is useful when the DNA molecule is longer, high-throughput methods have become more widely used, especially when entire genomes need to be sequenced.
There are three major changes compared to the Sanger method. The first was the development of a cell-free system for cloning DNA fragments. Traditionally, the stretch of DNA that needed to be sequenced was first cloned into a prokaryotic plasmid and amplified within bacteria before being extracted and purified. High throughput sequencing or next-generation sequencing technologies no longer relied on this labor-intensive and time-intensive procedure.
Secondly, these methods created space to run millions of sequencing reactions in parallel. This was a huge step forward from the initial methods where eight different reaction mixtures were needed to produce a single reliable nucleotide sequence. Finally, there is no separation between the elongation and detection steps. The bases are identified as the sequencing reaction proceeds. While HTS decreased cost and time, their ‘reads’ were relatively short. That is, in order to assemble an entire genome, intense computation is necessary.
The advent of HTS has vastly expanded the applications for genomics. DNA sequencing has now become an integral part of basic science, translational research, medical diagnostics, and forensics.
Uses of DNA Sequencing
Traditional, chain-termination technology and HTS methods are used for different applications today. Sanger sequencing is now used mostly for de novo initial sequencing of a DNA molecule to obtain the primary sequence data for an organism or gene. The relatively short ‘reads’ coming off an HTS reaction (30-400 base pairs compared to the nearly a thousand base pair ‘reads’ from Sanger sequencing methods) make it difficult to create the entire genome of an organism from HTS methods alone. Occasionally, Sanger sequencing is also needed to validate the results of HTS.
On the other hand, HTS allows the use of DNA sequencing to understand single-nucleotide polymorphisms – among the most common types of genetic variation within a population. This becomes important in evolutionary biology as well as in the detection of mutated genes that can result in disease. For instance, sequence variations in samples from lung adenocarcinoma allowed the detection of rare mutations associated with the disease. The chromatin binding sites for specific nuclear proteins can also be accurately identified using these methods
Overall, DNA sequencing is becoming an integral part of many different applications.
Diagnostics
Genome sequencing is particularly useful for identifying the causes of rare genetic disorders. While more than 7800 diseases are associated with a Mendelian inheritance pattern, less than 4000 of those diseases have been definitively linked to a specific gene or mutation. Early analysis of the exon-genome, or exome, consisting of all the expressed genes of an organism, showed promise in identifying the causal alleles for many inherited illnesses. In one particular case, sequencing the genome of a child suffering from a severe form of inflammatory bowel disease connected the illness to a mutation in a gene associated with inflammation – XIAP. While the patient initially showed multiple symptoms suggestive of an immune deficiency, a bone marrow transplant was recommended based on the results of DNA sequencing. The child subsequently recovered from the ailment.
In addition, HTS has been an important player in developing a greater understanding of tumors and cancers. Understanding the genetic basis of a tumor or cancer enables doctors to have an extra tool in their kit for making diagnostic decisions. The Cancer Genome Atlas and International Cancer Genome Consortium have sequenced a large number of tumors and demonstrated that these growths can vary vastly in terms of their mutational landscape. This has also given a better understanding of the kind of treatment options that are ideal for each patient. For instance, the sequencing of the breast cancer genome identified two genes – BRCA1 and BRCA2 – whose pathogenic variants have an enormous impact on the likelihood of developing breast cancer. People with some pathogenic alleles even choose to have preventive surgeries such as double mastectomies.
Molecular Biology
DNA sequencing is now an integral part of most biological laboratories. It is used to verify the results of cloning exercises to understand the effect of particular genes. HTS technologies are used to study variations in the genetic compositions of plasmids, bacteria, yeast, nematodes or even mammals used in laboratory experiments. For instance, a cell line derived from breast cancer tissue, called HeLa, is used in many laboratories around the world and was earlier considered as a reliable cell line representing human breast tissue. Recent sequencing results have demonstrated large variations in the genome of HeLa cells from different sources, thereby reducing their utility in cell and molecular biology.
DNA sequencing gives insight into the regulatory elements within the genome of every cell, and the variations in their activity in different cell types and individuals. For instance, a particular gene may be permanently turned off in some tissues, while being constitutively expressed in others. Similarly, those with susceptibility for a specific ailment may regulate a gene differently from those who are immune. These differences in the regulatory regions of DNA can be demonstrated through sequencing and can give insight into the basis for a phenotype.
Recent advances have even allowed individual laboratories to study structural variations in the human genome – an undertaking that needed global collaboration two decades ago.
Forensics
The ability to use low concentrations of DNA to obtain reliable sequencing reads has been extremely useful to the forensic scientist. In particular, the potential to sequence every DNA within a sample is attractive, especially since a crime scene often contains genetic material from multiple people. HTS is slowly being adopted in many forensics labs for human identification. In addition, recent advances allow forensic scientists to sequence the exome of a person after death, especially to determine the cause of death. For instance, death due to poisoning will show changes to the exome in affected organs. On the other hand, DNA sequencing can also determine that the deceased had a preexisting genetic ailment or predisposition. The challenges in this field include the development of extremely reliable analysis software, especially since the results of HTS cannot be manually examined.
Quiz